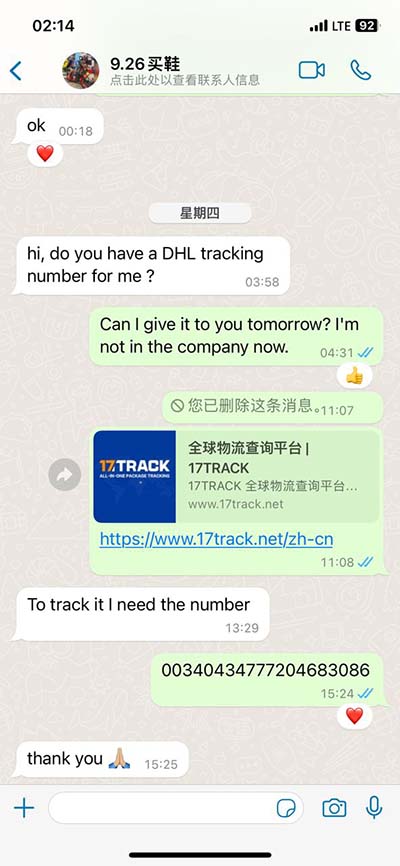
clustal omega online The original Clustal Omega tool for multiple protein sequence alignment. Clustal Omega is capable of aligning thousand of sequences and is an improvement of the previous version of .
This card can be Tribute Summoned with one Tribute using "Double Coston" and "The First Monarch" or tribute-free with "Sinister Yorishiro". Use "Mound of the Bound Creator" to protect this card and deal 1000 more Damage each time it destroys a monster by battle. "Magic Reflector" or "Field Barrier" can be used to prevent the Field Spell Card .
0 · multiple sequence alignment omega
1 · clustal online version
2 · clustal omega pairwise sequence alignment
3 · clustal omega job dispatcher
4 · clustal omega home page
5 · clustal omega guide tree
6 · clustal omega github
7 · clustal omega alignment tool
EX78 BossMonster Lv.88, LV.89. 시리즈입니다! 디자인, 투박하지만 중후한제품. 17인치 EX시리즈를 그대로 계승했습니다. 화려함보다는 엄숙함이 빛나는 외관이죠? 마찬가지로!! 8세대 커피레이크 데스크탑 CPU, i3-8100 그리고 i7-8700K 제품을 탑재! 더 높은 성능을 구사할 수 .
multiple sequence alignment omega
Clustal Omega is a new multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments between three or more sequences. For .Job Dispatcher. The Job Dispatcher at EMBL-EBI offers free access to a range of bioinformatics tools and biological datasets through its web and programmatic interfaces. It a.
clustal online version
Pairwise Alignment: FAST/APPROXIMATE SLOW/ACCURATE. Enter your sequences (with labels) below (copy & paste): PROTEIN DNA. Support Formats: FASTA (Pearson), .
Job Dispatcher. The Job Dispatcher at EMBL-EBI offers free access to a range of bioinformatics tools and biological datasets through its web and programmatic interfaces. It also powers .
Clustal Omega. Latest version of Clustal - fast and scalable (can align hundreds of thousands of sequences in hours), greater accuracy due to new HMM alignment engine.
The original Clustal Omega tool for multiple protein sequence alignment. Clustal Omega is capable of aligning thousand of sequences and is an improvement of the previous version of .
Clustal Omega is a multiple sequence alignment program for proteins. It produces biologically meaningful multiple sequence alignments of divergent sequences. Evolutionary relationships.Click on the Align link in the header bar to align two or more protein sequences with the Clustal Omega program. Enter either protein sequences in FASTA format or UniProt identifiers (as .
Clustal Omega [1] is a package for performing fast and accurate multiple sequence alignments (MSAs) of potentially large numbers of protein or DNA/RNA sequences. It is the .
clustal omega pairwise sequence alignment
Clustal Omega is a widely used package for carrying out multiple sequence alignment. Here, we describe some recent additions to the package and benchmark some alternative ways of making alignments. Clustal Omega [] is a package for performing fast and accurate multiple sequence alignments (MSAs) of potentially large numbers of protein or DNA/RNA sequences.It is the latest version of the popular and widely used Clustal MSA package [2, 3].Clustal Omega retains the basic progressive alignment MSA approach of the older ClustalX and ClustalW .Multiple Sequence Alignment by CLUSTALW: ETE3 MAFFT CLUSTALW PRRN; Help: General Setting Parameters: Output Format: Pairwise Alignment: FAST/APPROXIMATE SLOW/ACCURATE. Enter your sequences (with labels) below (copy & paste): PROTEIN DNA. Support Formats: FASTA (Pearson), NBRF/PIR, EMBL/Swiss Prot, GDE, CLUSTAL, and .Clustal Omega€is a multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments between€three or more€sequences.€It produces biologically meaningful multiple sequence alignments of .

Clustal Omega is a version, completely rewritten and revised in 2011, of the widely used Clustal series of programs for multiple sequence alignment. It can deal with very large numbers (many tens of thousands) of DNA/RNA or protein sequences due to its use of the mBed algorithm for calculating guide-trees. This algorithm allows very large . Clustal Omega is a package for making multiple sequence alignments of amino acid or nucleotide sequences, quickly and accurately. It is a complete upgrade and rewrite of earlier Clustal programs. This unit describes how to run Clustal Omega interactively from a command line, although it can also be run online from several sites. .homepage of the clustal series of programs (clustal omega, clustalw and clustalx) for multiple sequence alignment. Clustal W / Clustal X. Multiple alignment of nucleic acid and protein sequences Home; servers; . Instead, you can run Clustal online on several servers on the web: EBI web server Swiss Institute of Bioinformatics .
Clustal Omega uses a different method to calculate the guide tree compared to ClustalW, so we do not output the rough all-against-all pairwise alignment scores used to guide the alignment (as they don't exist). However we do calculate a pairwise identity matrix from the results, which can be downloaded from the Results Summary tab. .
The original Clustal Omega tool for multiple protein sequence alignment. Clustal Omega is capable of aligning thousand of sequences and is an improvement of the previous version of Clustal, ClustalW and ClustalX, using HMMs, based on HHalign from Johannes Soeding. Clustal Omega also makes use of precomputed aligment information found in public databases.
Clustal Omega is a package for making multiple sequence alignments of amino acid or nucleotide sequences, quickly and accurately. It is a complete upgrade and rewrite of earlier Clustal programs. This unit describes how to run Clustal Omega interactively from a command line, although it can also be run online from several sites. . Clustal Omega是欧洲生物信息研究所(EBI)开发的多序列比对排列工具,现已经完全取代了之前ClustalW的地位。最新本的omega比对准确度更高,而且速度更快,适合大规模的多序列比对。Clustal Omega is capable of aligning thousand of sequences and is an improvement of the previous version of Clustal, ClustalW and ClustalX, using HMMs, based on HHalign from Johannes Soeding. Clustal Omega also makes use of precomputed aligment information found in public databases. Clustal Omega data points are shown in red (default with a solid bullet, single linkage guide‐tree with a cross, maximum likelihood guide‐tree with a star, various iteration schemes with circles, itr1 and itr2 are single and double iterations). The remaining Clustal Omega data points correspond to options where guide‐tree and HMM .
Clustal Omega is a new multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments between three or more sequences. Link to EBI website. STEP 1 - Enter your input sequences. Enter or paste a set of amino acid sequences in any supported format: A multiple sequence alignment was prepared with Clustal Omega (Sievers and Higgins, 2021) using the EMBL-EBI online server and default parameters (Madeira et al., 2022). Aligned sequences were .
Clustal Omega data points are shown in red (default with a solid bullet, single linkage guide-tree with a cross, maximum likelihood guide-tree with a star, various iteration schemes with circles, itr1 and itr2 are single and double iterations). The remaining Clustal Omega data points correspond to options where guide-tree and HMM iterations are .Click on the Align link in the header bar to align two or more protein sequences with the Clustal Omega program; Enter either protein sequences in FASTA format or UniProt identifiers (as above) into the form field ; Click the ‘Run Align’ buttonClustal Omega also has powerful features for adding sequences to and exploiting information in existing alignments, making use of the vast amount of precomputed information in public databases like Pfam. Introduction. Multiple sequence alignments (MSAs) are essential in most bioinformatics analyses that involve comparing homologous sequences.In Clustal Omega, the alignments are then computed using the very accurate HHalign package (So¨ding, 2005), which aligns two profile hidden Markov models (Eddy, 1998). Clustal Omega has a number of features for adding sequences to existing alignments or for using existing alignments to help align new sequences. One innovation is
cartier hong kong website
Clustal Omega uses a different method to calculate the guide tree compared to ClustalW, so we do not output the rough all-against-all pairwise alignment scores used to guide the alignment (as they don't exist). However we do calculate a pairwise identity matrix from the results, which can be downloaded from the Results Summary tab. .
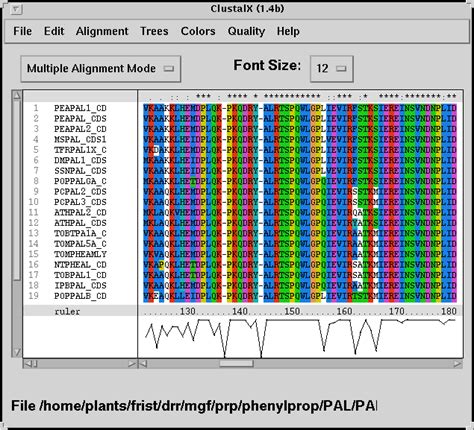
clustal omega job dispatcher
clustal omega home page
clustal omega guide tree
BMW e46 330d 135kw manuāls ļoti labā komplektācijā black sapphire krāsa. Rūpnīca. 2002. 3.0D. 350 tūkst. 3,600 €. Pārdodas ļoti labs luxury line bmw f31 330d xdrive ar .
clustal omega online|clustal omega home page